

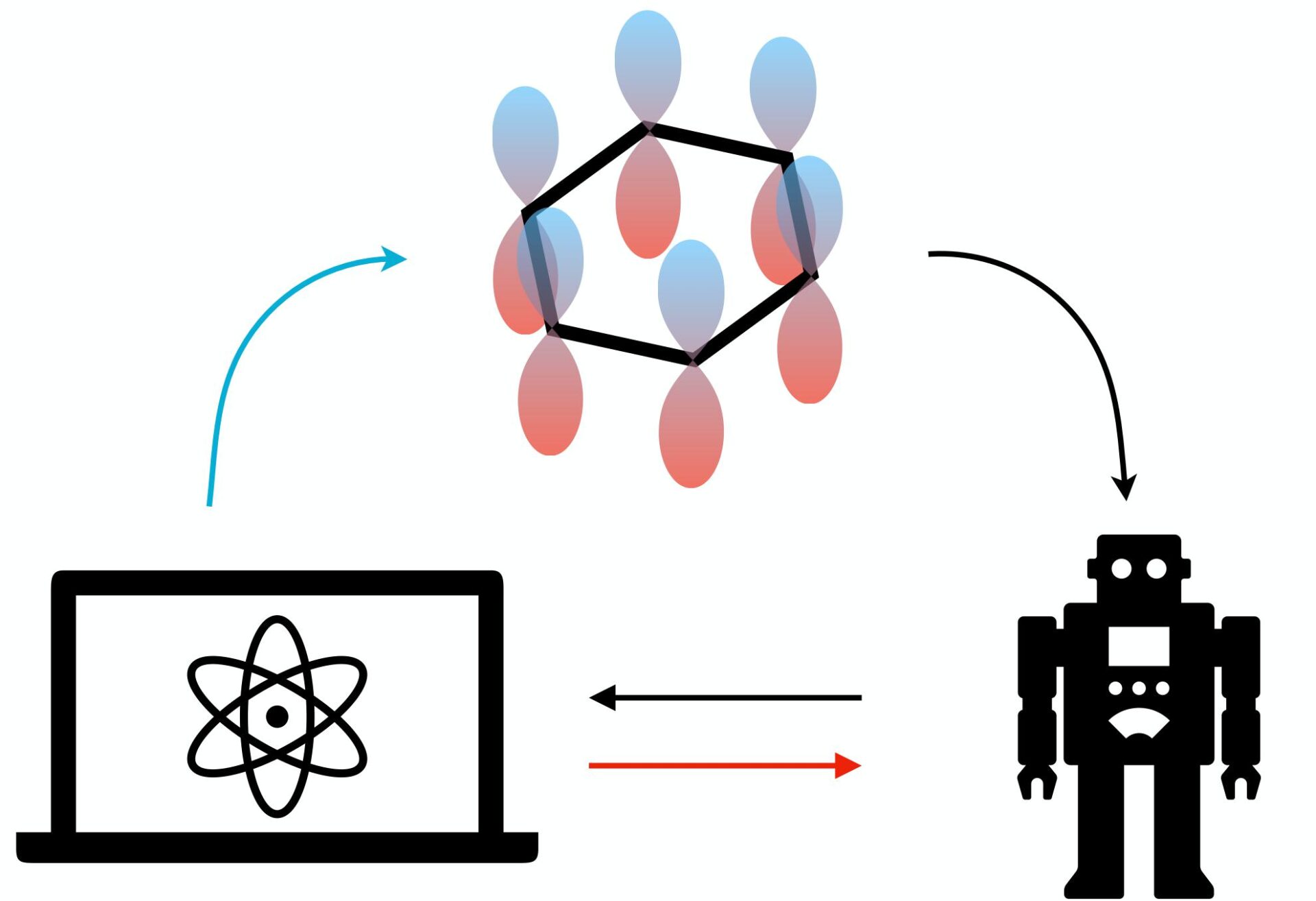
A new, interdisciplinary project combines expertise in machine learning, quantum algorithms, and chemistry to advance the abilities to calculate properties of molecules. In the long run, it could lead to speed-up of materials and drug design.
Properties of molecules, for example their energy levels, are important to know when designing new materials or drugs. However, these properties can only be calculated accurately for small molecules on conventional computers. For larger molecules, the computational problem becomes too large for the computer to handle. A solution could be to instead use quantum computers. Being quantum mechanical systems themselves, they are presumed to be good at simulating quantum mechanical systems like molecules. But it has turned out to be hard to figure out the right codes – so called quantum algorithms – to solve this task, as they comprise many parameters that must be optimised for the algorithm to work well. To tackle this challenge, the Wallenberg AI, Autonomous Systems and Software Program (WASP) and the Wallenberg Centre for Quantum Technology (WACQT) now launch a joint research project that combines expertise in machine learning, quantum computing, and chemistry.
“We will use an iterative cycle where a starting algorithm is run on a quantum computer. The output is then fed to a classical computer which uses machine learning to update the parameters of the quantum algorithm. This is repeated until we achieve a quantum algorithm which gives a good output,” explains Simon Olsson, assistant professor at the Department of Computer Science and Engineering at Chalmers and main principal investigator of the project.
His area of expertise is in machine learning applications in the sciences. Further principal investigators are Anton Frisk Kockum, quantum physicist and expert on iterative quantum algorithms, and Martin Rahm, chemist and expert on quantum algorithms in chemistry, both at Chalmers.
“The collaboration is a prerequisite for us to carry out this project really well,” says Frisk Kockum.
An ambition of the project is to use machine-learning algorithms to find very good starting guesses for algorithms to minimize the number of iterative cycles needed, as run-time on quantum computers is scarce and costly.
“We will start with simpler molecular systems and learn the relationship between the problem – for example to compute an energy – and the optimal quantum algorithm to get closer to the optimal programme from start – a ‘warm start’,” says Simon Olsson.
The project aims at demonstrating a proof of concept. In the long run, the work could contribute to speeding up materials and drug design, as well as optimisation problems, for example, in logistics.
In addition to the three principal investigators, the project will include a postdoc. An open position for a postdoc in the project is now available.
Read more
Published: February 7th, 2023
[addtoany]